BondVal
1.BondValの概要
BondValは結晶データから簡単にBond valence
sum(BVS)を計算するプログラムです。また、このプログラムは任意の原子間距離も標準偏差を伴って計算することもできます。
2.必要なファイル
BondVal.exe − 本体
BondVal.ini − 初期設定ファイル
bvparm2006.cif − r0を収録したデータベース(ソースファイルはここ)
spgra − 空間群データベース(Rietan2000に付属)(BondVal.iniの中で指定する)
ABCTrans − 格子の変換表(BondVal.iniの中で指定する)
BondVal.out − 出力ファイル
その他、種々のファイルが計算過程で出力されますが、全てテキストファイルですので中身を見れば大抵判断できます。
また、Rietan2000に付属のspgraもRietan2000のホームページから入手してください。
3.外部プログラム
計算結果を見るためにエディタを使用しています。使い慣れたエディタをBondVal.iniファイルの中に記述してください。
4.BondVal.iniの設定
・このファイルの前半部分に
[BondSum inifile]
SpgraFile = ..... :Rietan2000 付属のspgraファイル
SGTransFile = ..... :ABCTransファイル
StructView = ..... :拙作のStructView.exeがあるディレクトリ
の記述がある。各ファイルは絶対パスで指定すること。
・各種パラメータ
H-max = 2 :最近接原子(配位原子)を見つける際の隣接格子の範囲(変更することはまずない)
H-min = -2 :最近接原子(配位原子)を見つける際の隣接格子の範囲(変更することはまずない)
・editorはファイルの閲覧や作成に用いるので、自分にあった適当なエディタプログラムのファイル名を絶対パスで指定する。一般には
[Editor]
editor = C:\Windows\Notepad.exe
5.使い方
- BondValを立ち上げる。
- 結晶学的データを手動で入力する。必要項目は、タイトル名、空間群記号、空間群番号、空間群設定番号、格子定数、原子座標。原子座標のうち、[Species]の欄の入力は注意を要する。非対称単位に同じ原子種が複数ある場合には、元素記号の後にアルファベット以外の記号(例えば、数字)を付けること。
- 結晶学的データファイルを読み込める。読み込めるファイルは、
・BonVal専用のインプットファイル 例
-------------------------------------------------------------
# Title
Li Fe (P O4)
# Name of space group
P n m a
# Number and setting number of space group
62 1
# Unit cell parameters
10.32881 6.00340 4.69597 90.00000 90.00000 90.00000
0.00018 0.00011 0.00009 0.00000 0.00000 0.00000 <- 格子定数の標準偏差
# Number of sites
6
# Crystal data: name, g, x, y, z, charge, d-limit
Li 1.00000 0.00000 0.00000 0.00000 1.00000 2.2
0.00000 0.00000 0.00000 <- 部分座標の標準偏差
Fe 1.00000 0.28199 0.25000 0.97408 2.00000 2.8
0.00011 0.00000 0.00032 <- 部分座標の標準偏差
P 1.00000 0.09524 0.25000 0.41886 5.00000 2.0
0.00022 0.00000 0.00048 <- 部分座標の標準偏差
O(1) 1.00000 0.09766 0.25000 0.74472 -2.00000 3.0
0.00054 0.00000 0.00100 <- 部分座標の標準偏差
O(2) 1.00000 0.45509 0.25000 0.21498 -2.00000 3.0
0.00060 0.00000 0.00090 <- 部分座標の標準偏差
O(3) 1.00000 0.16632 0.04528 0.28035 -2.00000 3.0
0.00042 0.00060 0.00060 <- 部分座標の標準偏差
------------------------------------------------------------
(LatEnergyのインプットファイルとほぼ同じ。LatEnergyのインプットファイルも読み込める)
・Rietan2000のinsファイル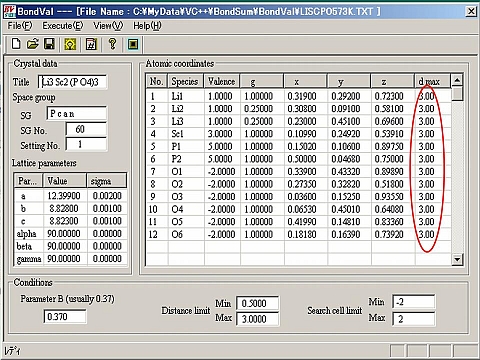
・Rietan2000のlstファイル
・ICSDファイル
・cifファイル(工事中)
注意:これらのファイルの読み込みに失敗した場合には、エディタでBonVal専用のインプットファイルを作成してください。
- 全原子に対する最近接原子間距離を求める際の範囲は、右図に示すように各原子の右端の欄(d-max)にその値を指定する(イオン半径が大きく異なるイオンを含む場合に有効)。
- データが正しく入力されたら、[Execute]->[Calculation]かTool
Buttomを押して計算を始める。
- 計算終了後、直ちにエディタが立ち上がり、結果が表示される(最初の図)。
6.アウトプットファイルの見方
- 原子間距離の部分
ファイルの中ほどに次の部分がある。
Searched distance:
No atom1 x y z atom2 site x y z d sigma(d)
1 Li 0.00000 0.00000 0.00000 O(1) 1_554 0.09766 0.25000 -0.25528 2.16959 0.00367
-- -- ------ ------ ------ ---- ----- ------ ------- ------ ------- ------
-- -- ------ ------ ------ ---- ----- ------ ------- ------ ------- ------
大体わかると思いますが、siteの意味は次の通り。最初の数値はこの上の部分に示されている対称操作の番号で、続く3つの数値はx,y,z,の順番に5を基準にして格子の平行移動の大きさを示す。この例(1_554)は1番目の操作で、z軸方向だけをマイナス側に1つ平行移動した原子(atom2)を示している。
- Bond valence sumの部分
各陽イオンについてBVSの値と配位数が記録される。無機化合物中の陽イオンの配位数は大体推定できるので、その値と異なっている場合には上述の6-4のところで述べたd-maxの値を変更して再び計算し直すことを勧める。
- BVSの値の下に計算に用いたr0の値が各結合ごとに出力される。最初のアルファベットはファイルの最後に示される文献に対応している。