まず、操作画面の一般的に使用する個所の説明をする。
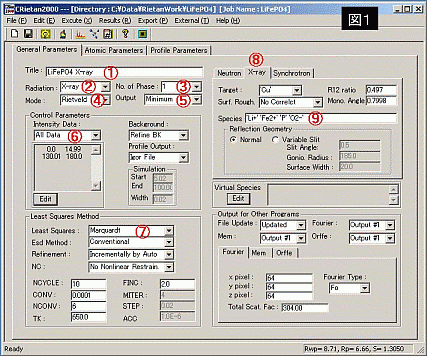
?@ 試料名 : 測定した試料の名前を入力する。
?A 相の数 : 単一相か二相か指定(ほとんどは"1"で良い。)
?B 測定線源 : 測定に使用した線源を指定する。
?C モード : 通常Rietveldで良い。(他にsimulation等もある。)
?D 結果の出力 : 通常、最小限の出力(minimum)で良い。
?E 計算除外範囲の指定 : 不純物ピークを計算から除外することができる。例えば、[1. not all data]で下の欄に20 25と入力すると20°から25°の範囲を除外できる。
?F 計算法の指定 : [Marquardt]がもっとも無難。 [Conjugate]は計算が遅く、精密化の最終段階で使用したほうがよい。
?G 測定線源の基礎データ指定 : 使用した線源の項目を選択し、波長等を適切に入力する。(普通選択するだけで良い)
?H 元素指定 : 測定した試料の元素を入力する。'○'で囲んで、元素名と酸化数を並べて入力(ただし、S,P,N等は価数なし) この価数は、[Rietan2000-folder]→[program]中の[asfdc]ファイルをテキスト等で開けば、確認できます。
図1右下部分 [Ourput for other programs] 補足プログラム
・[file update]: 他のプログラムファイルをアップデートするかどうか
・[Fourier syn]: フーリエ合成・D合成(差フーリエ合成)、結晶構造因子の逆フーリエ合成により、電子密度分布を計算する。ここでは、観測した結晶構造因子と理論値の結晶構造因子との差からフーリエ合成する。(プログラム名:Fousyn)
・[Mem output]: 最大エントロピー法(maximum-entropy method)、これも[Fourier syn]同様、電子分布密度の計算を行う。(プログラム名:Prima)
・[Orffe output]: 原子座標から、原子間距離の計算を行う。普通、[file update]とともに、作動するようにしておく。
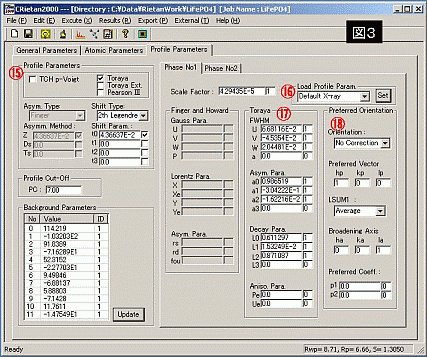
?I 測定試料名 : '○'で必ず囲む
?J 空間群 : インターナショナルテーブルにより、試料の空間群を探し、入力する。monoclinic等、軸の取り方により設定が変わる化合物はその番号も付与する。(例:A-14-5)
?K 原子数 : 解析に使用する原子の数を入力する。(下表の原子数と同じ数値を入力)
?L データ読みこみ : ICSD等のテキストファイルから原子座標データ等を読み込むことができる。これを使用すると、このページは自動で書きこむことができる。(Find Itからの場合、Longファイルを.txtで保存し、そのファイルを読み込むと良い)
?M 原子の座標、パラメータ : 原子の座標、パラメータを入力する。
(ICSDで読みこんだら、原子の価数に注意する。図1の?Hに入力した化学種と同じにする)
・occupancy: 占有率、完全充填は1、全く占有していない場合0、1を超えてはならず、負にもならない。
・x, y, z: 原子座標
・B: 等方性熱振動パラメータ
それぞれの数値の隣にある0, 1といった数値は変動制御部で0は固定、1は変動を表す。