Ⅰ セットアップの仕方
ご利用している方々からCRietan2000が上手く走らないとの苦情が多数ありましたので、今回からはセットアッププログラムをなくすことにしました。
セットアップの仕方はいたって簡単で、CRietan2000.lzhを必ずC:ドライブのルートディレクトリに解凍してください。CRietan2000.lzhの中はディレクトリ構造を保持していますので、解凍すれば必ずC:\CRietan2000のフォルダができます。Rietan2000については、泉氏のホームページ
からrietan2000w.tbzをダウンロードし、これをCRietan2000のディレクトリに解凍する。このディレクトリの下に"RIETAN-2000 Folder"ディレクトリが作られます。
ディレクトリ構造は以下のようになります。
c:\CRietan2000----Cell(*)
|
|---DATA
|
|---PROG
|
|------------CellRef(*)
|------------Database
|------------Doc
|
|---CRietan.files
|
|------------Example
|------------EXPO(*)
|------------FourierMap
|------------GenHKL(*)
|------------PatView
|
|----PatView.files
|
|------------RIETAN-2000
Folder(*)
| |---batch
files
| |---lst2cif
| |---orffe
| |---proprams
| |---templates
| |---VENUS
|
|------------StructView
|------------Valist(*)
(*)印の付いているディレクトリはCRietan2000.lzhのなかには含まれていませんので各自用意してください。
[上記以外のディレクトリにセットアップしたい場合]
1.CRietan2000をインストールしたディレクトリにあるCRietan2000.iniファイルの前半部分に下記の設定を書く。
SET
RIETAN=%Rietan%\RIETAN-2000 Folder\programs
SET
ORFFE=%Rietan%\RIETAN-2000 Folder\programs
SET
DATABASE=%Rietan%\RIETAN-2000 Folder\programs
SET
LST2CIF=%Rietan%\RIETAN-2000 Folder\programs
SET
PRIMA=%Rietan%\RIETAN-2000 Folder\VENUS\program\
SET
VEND=%Rietan%\RIETAN-2000 Folder\VENUS\programs
SET
VICS=%Rietan%\RIETAN-2000
Folder\VENUS\programs
ここで、%Rietan%はRietan2000をインストールしたディレクトリを示す。例えば、"c:\Chemisty"など。
注意:CRietan2000をインストールするディレクトリの名前には、日本語名や空白のある名前はできるだけ避けること。また、あまり長い名前も好ましくない。
2.StructViewディレクトリにあるStructView.iniファイルを最後にある[Database]の指定を書き換える。
Ⅱ Rietan用X線データ(.intファイル)を作る。
(ⅰ) [CRietan2000]を起動する。新規に解析を行う場合は適当なフォルダを指定してから、ジョブ名を入力する。
このジョブ名によって以後いろいろなファイルが作られる。解析途中の場合は当該フォルダに移動してから目的(適当な)の○.insファイルを開く。下の図の[brouse]を押すとファイル指定がされる。ファイルがない場合、[cancel]を押すとdefaultの設定が読みこまれる。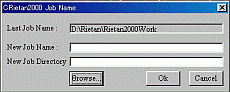
(ⅱ) メイン画面が表示されたら、[File]の[make int file]を選択する。(pattern viewが開く)
(ⅲ) Pat View上の[File]中の[Load raw data]から、必要なデータ方式を選ぶ
[Rigaku Rin
Data] 理学電機製Rintシステムのデータ
[Rigaku Old Format Data] 古い理学電機製システムのデータ
[Mac
Data] 本学化学システム工学科の佐藤峰夫研究室が所持するX線回折装置のデータ
[Hermes Nuetron
Data] 東北大学金研が所持する中性子回折装置のデータ
[(x,y) Format
Data] (2θ,Intensity)の並びのデータ
(ⅳ) [File]→[Export powder formats]→[Rietan normal
format]を選び、[1step obs]を選択、
[○.int]ファイルでセーブする。(○は試料名などを入れる。ここで試料フォルダを作成し、次に作成するinsファイルと一緒にそのフォルダに保存する。)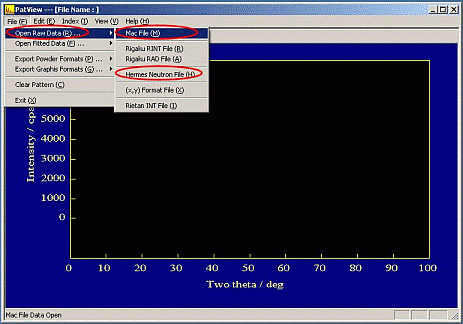
<他の方法によるintファイルの作成>
上述した方法では、[1step
obs]を選択して保存したが、これは、X線プロファイルのバックグランドが小さく、ファイルの大きさも小さい場合である。 バックグラウンドが大きいとRietveld解析において、正確な精密化が困難になる。また、データが大きいと処理時間が長く、計算に失敗する可能性も出てくるので、この回避法を示す。まず、上述した(ⅰ)、(ⅱ)、(ⅲ)の方法で[Pat
View]を起動する。そして、X線プロファイルを表示する。
① バックグラウンドが大きい場合
[pattern]→[smoothing]でスムージングする。(大体、3ポイントごとで良い)
[File]→[Export
powder formats]→[Rietan normal format]→[1steps
calc](1ステップ毎の計算値)を選び、[○.int]ファイルでセーブする。
② プロファイルデータが大きい場合
[File]→[Export powder formats]→[Rietan normal
format]→[2steps obs](2ステップ毎の実測値)を選び、[○.int]ファイルでセーブする。
③ バックグラウンド、データともに大きい場合、①と②を複合してintファイルを作成する。
Ⅲ リータンの初期設定(.insファイルの作り方)
まず、操作画面の一般的に使用する個所の説明をする。
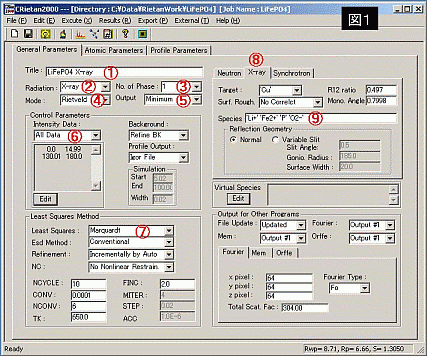 ① 試料名 : 測定した試料の名前を入力する。
① 試料名 : 測定した試料の名前を入力する。
② 相の数 : 単一相か二相か指定(ほとんどは"1"で良い。)
③ 測定線源 : 測定に使用した線源を指定する。
④ モード : 通常Rietveldで良い。(他にsimulation等もある。)
⑤ 結果の出力 : 通常、最小限の出力(minimum)で良い。
⑥ 計算除外範囲の指定 : 不純物ピークを計算から除外することができる。例えば、[1. not all
data]で下の欄に「20 25」と入力すると20°から25°の範囲を除外できる。
⑦ 計算法の指定 : [Marquardt]がもっとも無難。
[Conjugate]は計算が遅く、精密化の最終段階で使用したほうがよい。
⑧ 測定線源の基礎データ指定 : 使用した線源の項目を選択し、波長等を適切に入力する。(普通選択するだけで良い)
⑨ 元素指定 : 測定した試料の元素を入力する。'○'で囲んで、元素名と酸化数を並べて入力(ただし、S,P,N等は価数なし)。 この価数は、[Rietan2000-folder]→[program]中の[asfdc]ファイルをテキスト等で開けば、確認できます。
図1右下部分 [Ourput for other
programs] 補足プログラム
・[file
update]: insファイルをアップデートするかどうか
・[Fourier
syn]: フーリエ合成・D合成(差フーリエ合成)、結晶構造因子の逆フーリエ合成により、電子密度分布を計算する。ここでは、観測した結晶構造因子と理論値の結晶構造因子との差からフーリエ合成する。(プログラム名:Fousyn)
・[Mem
output]:最大エントロピー法(maximum-entropy method)、これも[Fourier
syn]同様、電子分布密度の計算を行う。(プログラム名:Prima)
・[Orffe output]:原子座標から、原子間距離の計算を行う。普通、[file
update]とともに、作動するようにしておく。
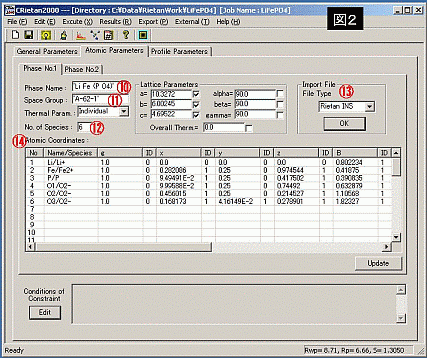
⑩ 測定試料名 : ' ' で必ず囲む
⑪ 空間群 : インターナショナルテーブルにより、試料の空間群を探し、入力する。monoclinic等、軸の取り方により設定が変わる化合物はその番号も付与する。(例:A-14-5)
⑫ 原子数 : 解析に使用する原子の数を入力する。(下表の原子数と同じ数値を入力)
⑬ データ読みこみ : ICSD等のテキストファイルから原子座標データ等を読み込むことができる。これを使用すると、このページは自動で書きこむことができる。(Find Itからの場合、Longファイルを.txtで保存し、そのファイルを読み込むと良い)
⑭ 原子の座標、パラメータ : 原子の座標、パラメータを入力する。(ICSDで読みこんだら、原子の価数に注意する。図1の⑨に入力した化学種と同じにする)
occupancy: 占有率、完全充填は1、全く占有していない場合0、1を超えてはならず、負にもならない。
x,
y, z: 原子座標
B: 等方性熱振動パラメータ
それぞれの数値の隣にある0, 1といった数値は変動制御部で0は固定、1は変動を表す。
⑮ プロファイル関数の指定 : たいていの場合、[Toraya]でOK。[TCH p-Voigt]はFinger法とHaward法の2種類ある。下のパラメータは大抵デフォルトのままでよい。
⑯ デフォルト値読みこみ : 精密化の途中で収束できなくなった場合などに、測定法を選んでこの[set]ボタンを押してプロファイルパラメータをデフォルト値にすると上手くいくことがある。
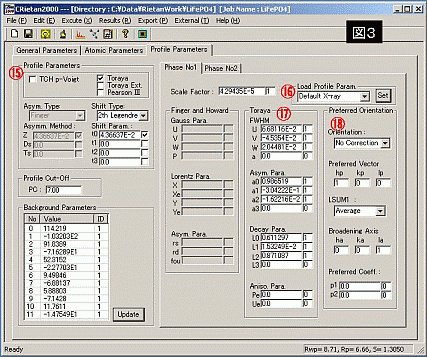
⑰ 変数 : プロファイル関数の各種の変数を表示。⑮の関数を変更すると、これらのパラメータも自動的に切り替わる。個々の変数の意味はRietan2000のマニュアルを参照のこと。
⑱ 配向制御 : 配向のある試料の場合に使用する。 (設定しない場合、[Orientation]を[not refined]とする。)
・[needle]: 針状結晶
・[plate]: 板状結晶
・[mulch-dollase]: すべての結晶
[proferred
Vector]で配向面を設定。[preferred coeffient]の[p1](表面粗さパラメーター)を1.0, 1にすると、選択配向が有効になる。
● insファイルの作成例(X線の場合)
① 試料名を記入(合成条件等も書くとわかりやすい)
② "1"にする
③ "X-ray"を選択
④ "Rietveld"を選択
⑤ "minimum"を選択
⑥ "All
data"を選択
⑦ "marquard[Marquardt]"を選択
⑧ "X-ray"を選択
⑨ 元素を指定
⑬ icsdデータの読みこみ。一覧から[ICSD]を選択し、[open
xyz file]を押し、ファイルを選択する。
始めは、[lattice parameters]のチェックを晶系に合わせてはずす。
⑭ [Atomic
coodinates]の元素価数を図1で設定した値と同じに指定し、occupancy, x, y, z,
Bそれぞれの隣にある横幅の小さい欄の数値をすべて0にする。
⑯ [Update]を押す 。(忘れることがよくある
)
以上の数値等を設定したら、[file]→[save as
ins file]でinsファイルを保存する。
(save as ...だと別名保存が可能)
<補足>
ここに掲載した図では、[General Para]のページの右下部分が[Output
#1]となっているが、これは、フーリエ合成、MEM計算、原子間距離の計算をする場合に使用する。フーリエマップの作成法はⅥで説明。
Ⅳ 精密化の順序
(ⅰ) insファイルを保存したら、まず、上部にある (RUNボタン)を押す。
(RUNボタン)を押す。
(MS‐DOS画面が表示されて、計算が進んでいることが確認されます。)
(ⅱ) 計算がcompleteしたら、テキストエディタが開いて計算結果が表示される。また、下部のR値(信頼値)も表示される。Rietan2000の解析結果は上部の (テキストのボタン)を押せばいつでも確認できる。 (出力テキストの真中あたりのncycleの表記のところでover~と言った表示がなければ良い。また、最後の方にある元素分率を計算してみて理論値と合っていれば良い。)
(テキストのボタン)を押せばいつでも確認できる。 (出力テキストの真中あたりのncycleの表記のところでover~と言った表示がなければ良い。また、最後の方にある元素分率を計算してみて理論値と合っていれば良い。)
もし、failed(失敗)していても、このテキストを確認すれば、何が原因で失敗したのか分かる。(FWHMと表示されたら、[Profile
Para]のFWHMの部分の数値を1~2桁下げてやると動くときがある。)
(ⅲ) 計算が成功したら、上部の (Updateボタン)を押して、アップデートする。 (Rwp値があまり下がらない(30~)場合、もう一度、RUNしてみると良い・・・かも) アップデートして、値がおかしいとかRUNで失敗する等の障害が出て、ひとつ前に戻したい時は、
insファイルが保存されているフォルダ中に「○.$$$」ファイルが存在するのでこれの$$$部分をinsに変換して、読み込むとひとつ前の状態に戻る。
(Updateボタン)を押して、アップデートする。 (Rwp値があまり下がらない(30~)場合、もう一度、RUNしてみると良い・・・かも) アップデートして、値がおかしいとかRUNで失敗する等の障害が出て、ひとつ前に戻したい時は、
insファイルが保存されているフォルダ中に「○.$$$」ファイルが存在するのでこれの$$$部分をinsに変換して、読み込むとひとつ前の状態に戻る。
以上の操作を2~3回繰り返し、Rwp値があまり変化しなくなったら、次に進む。
(ⅳ) [Atomic Parameter](図2)の[lattice
parameters]の変化する部分(最初の状態)をチェック。⑭のoccupancy, x, y, z,
Bそれぞれの隣にある部分の0を1に変える(必要部分のみ)。
occupancy: 占有率、変化する場合のみ1にする。→占有率等を固定、共有する場合
x,
y,
z: インターナショナルテーブルの特殊(等価)位置は0にする。他の一般位置は1で良い。
B: ほとんどの元素について初期値は0.5で良い。ただし、X線測定での場合、Li等の軽元素は測定できないので、0.5などに固定する。 [Profile
Para](図3)のバックグラウンドパラメータの右側部分をすべて0にする。→RUN
(ⅴ) 上部 でグラフ、テキストを確認してから、[update] → RUNを繰り返す。<注意>update後に[Atomic
Para](図2)の熱振動パラメータ(Beq)がマイナスもしくは1以上の場合、0.5に書きなおしから、RUNすること。
でグラフ、テキストを確認してから、[update] → RUNを繰り返す。<注意>update後に[Atomic
Para](図2)の熱振動パラメータ(Beq)がマイナスもしくは1以上の場合、0.5に書きなおしから、RUNすること。
(ⅵ) ある程度、R値が下がったら、[Profile
Para](図3)のバックグラウンドパラメータの右側をすべて(1~10)1にして、⑦の部分を[conjugate]にしてRUNする。(conjugateの回転数は3~4回で良い。)
(ⅶ) Rwp値が10近くもしくは10以下なら、終了。
Rwp,
Rp: 回折強度やバックグラウンド強度を含むパターンを対象とするR 因子
RI,
RF: それぞれ積分強度と構造因子を対象とするR 因子
S(テキストの中盤あたりに表記): =Rwp/Rp フィットの良さを示す。S=1は精密化が完璧なことを示す。普通、1.3より小さければ、満足する解析結果と言える。ただし、[Excute] → [Run
orffe] → [Result] → [Show DST file]で原子間距離、配位を確認し、理論値に近ければ、成功である。
([General Para](図1)のページの右下部分の[Orffe
output]が[Update~]となっていなければならない。)
Ⅳ-2 占有率等の変数の計算設定の仕方
ここでは、
●二つの元素が一つのサイトを共有している。
●同一元素が異なるサイトにあり、占有率が分配されている。
●予測挿入サイトが多くあり、元素挿入の分配の可能性がある場合
●二相共存(異なる物質が混合)での解析
等の計算設定を行う際の操作方法を説明する。
逆スピネル構造を持つZnMgSnO4について解析した例を説明する。この物質はすでに報告されている同じ逆スピネル構造を持つZn2SnO4とMg2SnO4との固溶体とみなせる。空間群はFd-3m(227)、Z=8である。この固溶体では、(Zn1-xMgx)t[ZnxMg1-xSn]oO4として表すことができるとした。ここで、( )tは四面体の8aサイト、[ ]oは八面体の16dサイトを表す。したがって、8aサイトには2種類、16dサイトには3種類の元素が存在する。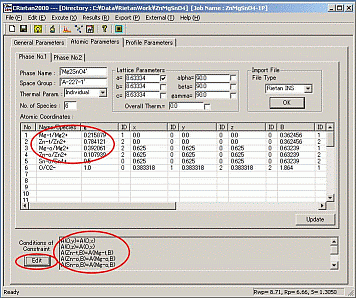
① ICSDデータベースにあるZn2SnO4とMg2SnO4との座標値を参考にして[Atomic
Parameter]の構造パラメータの初期値を作成する。ここでは、8aサイトにある元素をZn-tとMg-t、16dサイトのそれをZn-o、Mg-o、Sn-oとしている。
② 最初の計算段階では、これらの化学種の席占有率を全て0.5で固定し、さらに熱振動パラメータも0.5に固定して計算する。
③ その後、Zn-t、Mg-t、Zn-o、Mg-oの席占有率を可変(変動制御部で1を入力する)、また熱振動パラメータの全てを可変にする。
④ 追加された二つのLiサイトの占有率の和が1なのでこれを考慮した計算をしなければならない。ここで、(Zn1-xMgx)t[ZnxMg1-xSn]oO4の組成式より、Mg-tの席占有率をgとするとそれぞれの化学種の席占有率は、
Mg-t -> gx(Z/8)=g
Zn-t -> (1.0-g)x(Z/8)=1.0-1.0xg
Mg-o -> (1.0-g)x(Z/16)=0.5-0.5xg
Zn-o -> gx(Z/16)=0.5xg
Mg-t -> 1x(Z/16)=
0.5
である。ここで、式中の8と16は8aと16dに対応している。そこで、これらの関係式の入力を[Atomic
parameters]のCondition of
Constraintの[Edit]で使って行う。下の図では、既に入力が終わっているが、最下部のテキスト欄に条件式が記述されているのがわかる。
⑤ [Edit]ボタンを押すと下の図のウインドウが表示される。 ここでは、Mg-tの席占有率が精密化されるとしている。
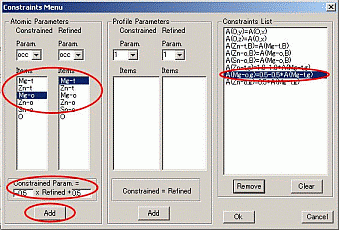
このウィンドウでの手順
(1) RefinedのParam.として[occ]を選択し、その下の[Items]のMg-tを選択する。
(2) ConstrainedのParam.として[occ]を選択し、その下の[Items]にあるZn-t、Mg-o、Zn-oについて前の式に従って条件を設定する。ここでは、Mg-oについて具体的に見てみよう。
・[Items]としてMg-oを選択する。
・Mg-o -> (1.0-g)x(Z/16)=0.5-0.5xgの式より、下にある[Constrained
Param.=]のテキスト欄に-0.5と0.5を入力する。
・その下の[Add]ボタンを押すと [Constraints
List]に条件式が示される。
(3) 他の2つの化学種についても同様に条件を入力する。
(4) 熱振動パラメータの制約条件については、Zn-tとMg-tのB、およびZn-o、Mg-o、Sn-oを共通すにする必要がある。この場合、Param.としてBを選択して、[Constrained
Param.=]のテキスト欄には1.0と0.0を入力すればよい。([Constraints
List]参照)
(5) またこの例では、酸素は32eの特殊位置(x,x,x)にあるのでOのyとz座標はx座標と共通にしている。この条件の入力の仕方はご自分で試していただきたい。
⑥ 最後に[Ok]ボタンを押すとこのWindowが閉じて、その結果が④の最下部のテキスト欄に反映されるとともに、制約条件が課されたパラメータのID欄に2が入力される。
● 二相共存で行う場合
[File]→[Import Template File]→[Two
Phase X-ray file](X線データの場合)か[Two Phase Neutron
File](中性子回折の場合)を選択し、デフォルト値を読み込ませてから、必要な値や設定を行い、精密化すると良い。
Ⅴ 格子ひずみと結晶子サイズの解析
 ※この操作は普通の操作を行い、Rwpが十分に下がった状態からすること
※この操作は普通の操作を行い、Rwpが十分に下がった状態からすること
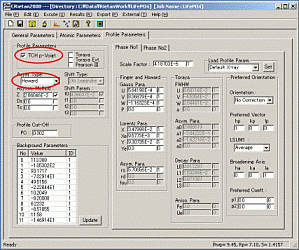
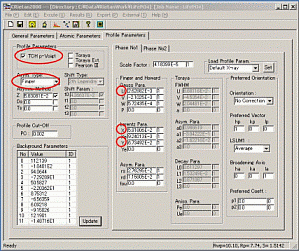
(ⅰ) [Profile Parameter]の[Common Profile
Parameters]で[TCH
p-Voigt]を選択する。
(ⅱ) [Asym.
Type]を[Howard]にする。
(ⅲ) [General
parameters]の左下の計算方法を[Conjugate]にして、Run。
(ⅳ) update後、[Asym.
Type]を[Finger]にして、Run。([Conjugate]でRwpが十分に下がるまで計算する。)
注:この計算は時間が長くかかり、計算中に発散しやすい。計算後には、原子間距離等の確認を行うこと。
(ⅳ) [Profile
Parameters]の[Finger]の値を使用して計算をする。
ここでは、[Howard]法→[Finger]法と順番に行ったが、発散する可能性が出てくるため、
[Howard]法、[Finger]法どちらか一方のみで精密化を行っても良い。
○結晶子サイズ(p)の計算 (X線プロファイルの半値幅により計算される。)
注:結晶子サイズの単位はX線の波長の単位と連動する。
p = 180Kλ/πX (nm)
K: シェラー定数(=
0.9)
λ: X線の波長(nm)
π: 円周率
X: Lorentz Para(対称プロファイルパラメータ)
○格子ひずみ(S)の計算
ガウス関数(U: Gauss Para)による
SG =
(π/180)(8ln2U)1/2×100 (%)
ローレンツ関数(Y: Lorentz Para)による
SL =
(π/180)Y×100 (%)
Ⅵ フーリエ解析による原子密度分布
※この操作は普通の操作を行い、Rwpが十分に下がった状態からすること。(計算した直後でないと作動しない。)
① フーリエ解析
フーリエ解析では測定されていない高次反射の打ち切り効果により、幻のピークや負の電子密度が出現しやすい。
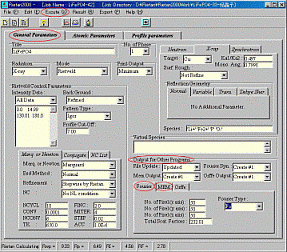
(1) [General Parameter]の[Output for other
programs]で[fourier]で[Output #1]を選択する。
(2) 下のTabで[Fourier]を選択して、ピクセル数を入力する。これは格子定数の大きさを参考にして適当な数値を入れること。
(3) [Fourier Type]を選択する。
(4) [Total
Scat. Fact.]とは、単位格子中の電子の総和である。(計算式あり)
(5) これらの入力を確認してから、一度[Run]を実行する。
(6) [Execute]の[Run fousyn]を選択し、実行すると、下記のダイアログが現れる。大抵の場合、「はい」でOk。
(7) fousyn.exeよる計算が始まる。 
(8) 計算が終了したらエディタが立ち上がり計算結果が表示される。数値ばかりで見てもつまらない。
(9) 成功したら、[Execute]→[Fourier
Map]を選択すると、下図のマップが現れる。
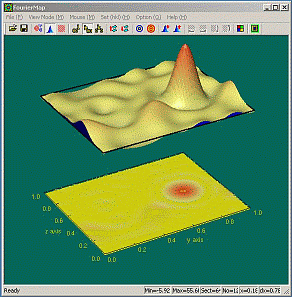
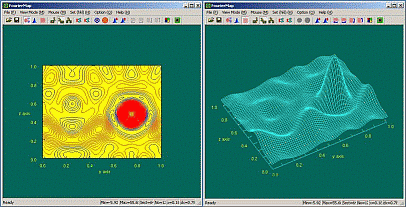
このFourierMap.exeにはいろいろな機能がある。例えば、マップの種類として上記の3Dマップの他に2Dマップ(コンターマップ)とメッシュマップがある。種々のコマンドボタンがあるので試していただきたい。
② MEM解析
MEM解析についてもフーリエ解析ほぼ同様にして行える。実行は[Execute]の[Run
Prima]で行い、解析結果はExecute]の[Prima Map](実際はVend.exe)で見ることができる.。実際に行うには、かなり面倒な手続きを経るので、Primaのマニュアルをよく読んでから行うことをお勧めする。
左図はPrimaのprfファイルの編集、右図はPrimaの実行画面を示す。
左図はPrimaの計算結果、右図はVENDで描いた電子密度をそれぞれ表す。
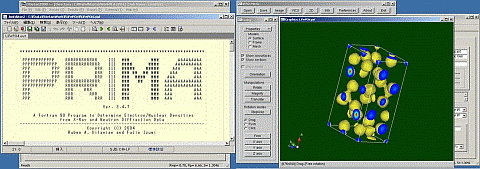
Ⅶ 結晶構造の示し方
ここでは、プログラム「Atoms」を使用して、結晶構造を示す方法を示します。
① 精密化を行う。(計算を行った直後に以下の操作を行うこと)
② 上部の[Export]→[Shelx ins
file]を押す。(これで、同一フォルダに[shelx.ins]ファイルが生成される。)
③ Atomsを起動する。
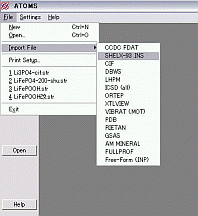
[File]→[import
file]→[Shelx93.ins]と押す。
下の図のウインドウが表示されるのでここでOKし、目的試料のフォルダから[shelx.ins]を読み込む。「Calculate
now?」と聞かれるので、OKする。すると、原子の位置が三次元的に表記される。
この後、ポリヘドラ等の設定を行うのだが、これは、Manual
Topページにある 「Atoms」の使用法を参考にしてください。
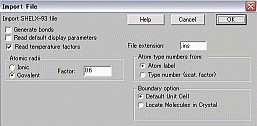
Ⅷ 画像(Fitting pattern)出力の仕方
解析が終了して、報告書やOHP、パワーポイント等にパターンを貼り付けたい場合、以下の操作を行います。
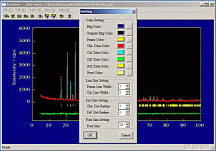
① 上部のを押し、[Pat
View]を起動する。
② [View]→[Set
Para]で色、フォントの大きさ等を指定する。
③ [File]→[Export Graphic Formats]→[EMF
Format]でBlack&WhiteかColorを選択する。そして、画像ファイルの保存先を指定し、名前をつけて保存する。[Sma4]はグラフ作成ソフト(シェアウェア)Sma4用の入力ファイルを出力する。
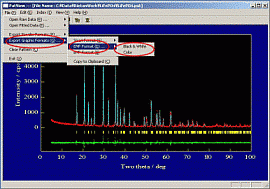 ④ Wordを起動し、[挿入]→[図]でファイルを指定すれば、フィッティングパターンが貼り付けられます。
④ Wordを起動し、[挿入]→[図]でファイルを指定すれば、フィッティングパターンが貼り付けられます。
bmpファイルとemfファイルは相当大きなファイルとなるので、Sma4を使って図を作ったほうが良いかもしれない。
戻る