-
LatEnergyを立ち上げる。
-
結晶学的データを手動で入力する。必要項目は、タイトル名、空間群記号、空間群番号、空間群設定番号、格子定数、原子座標。原子座標のうち、[Species]の欄の入力は注意を要する。非対称単位に同じ原子種が複数ある場合には、元素記号の後にアルファベット以外の記号(例えば、数字)を付けること。
-
結晶学的データファイルを読み込める。読み込めるファイルは、
・LatEnergy専用のインプットファイル 例
-------------------------------------------------------------
# Title
Li Fe (P O4)
# Name of space group
P n m a
# Number and setting number of space group
62 1
# Unit cell parameters
10.32680 6.00228 4.69507 90.00000 90.00000 90.00000
# Number of sites
6
# Crystal data: name, g, x, y, z, charge
Li 1.00000 0.00000 0.00000 0.00000 1.00000
Fe 1.00000 0.28201 0.25000 0.97396 2.00000
P 1.00000 0.09528 0.25000 0.41846 5.00000
O(1) 1.00000 0.09790 0.25000 0.74515 -2.00000
O(2) 1.00000 0.45516 0.25000 0.21210 -2.00000
O(3) 1.00000 0.16681 0.04638 0.28011 -2.00000
------------------------------------------------------------
・Rietan2000のinsファイル
・Rietan2000のlstファイル
・ICSDファイル
・cifファイル(工事中)
注意:これらのファイルの読み込みに失敗した場合には、手動で入力してください。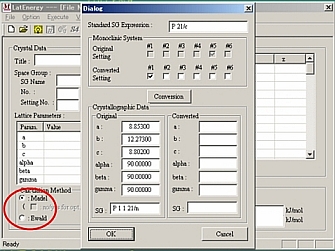
-
結晶学的データファイルを読み込んだ際に、種々のメッセージが現れることがある。単斜晶系と斜方晶系の場合、軸の変換を促すウィンドウが現れる。共に6種類の変換法がある。単斜晶系の場合どれを選んでもよいが、角度の1つが150°を超えるような変換ではうまくいかないので注意。また、斜方晶系の場合には変換軸は標準設定を選ばなければいけないので、必ずNo.1(abc)の設定を選択すること。
-
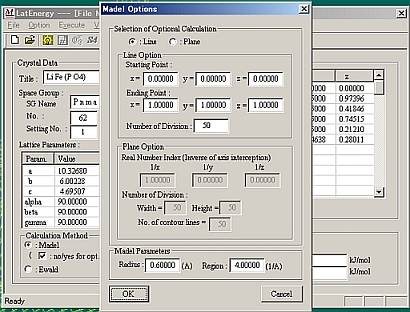
[Calculation Method]の中のRadio ボタンで計算方法を選択する。Madel法では、オプションをチェックするとオプションウィンドウが開くので、LineかPlaneを選択する。Planeオプションでは、x、y、z軸のどれかに平行な面を指定できる。
-
全ての項目を正しく入力し終わったら、[Execute]->[Calculate]を実行すると、計算ウィンドウが表示され、計算が始まる。計算が終わり、ウィンドウを閉じるとCalculation Results欄に結果が表示される。詳細な出力を見たい場合には、[Execute]->[Show Calculate Results]を実行して確認できる。全電荷の合計がゼロにならないときにはその旨が記録される。
「**Warning ! Total charge sum is not equal zero. ChargeSum = 24.00」など。この場合、元素の電荷をチェックすることを勧める。
-
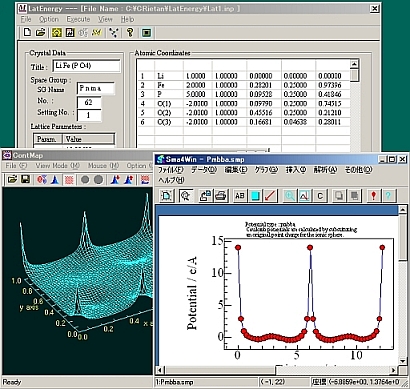
Lineオプションを指定した場合、[View]->[Sma4]->[Pmbba] or [Pobba]でSma4が立ち上がり、即座にグラフが描かれる。PmbbaとPobbaについてはMadelのマニュアルを参照して下さい。
-
Planeオプションを指定した場合、[Execute]->[Contour Map]でポテンシャル面が表示できる。この図は適当な画像ファイルに保存できる。(冒頭に示した図)
-
Ewald法を指定した場合、詳細出力には、近接反発ポテンシャルを含めた格子エネルギーが出力されているが、この値は、近接反発ポテンシャルとして(bi+bj)exp((ai+bi-rij)/(bi+bj))の形を仮定して求めている。ただし、各元素のai、bi、rijパラメータの値(MDParaファイルにある)はかなり適当な値であるのであまり信用できないと思われる(^_^;)。
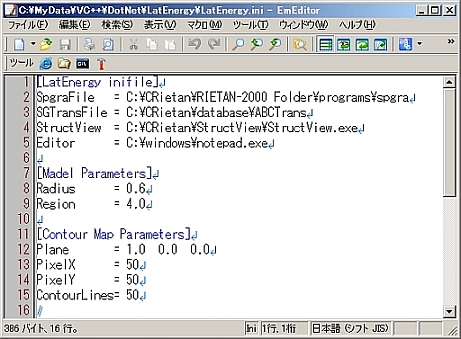 4.LatEnergy.iniの設定
4.LatEnergy.iniの設定