5.1 Manual Search
回折データを読み込んだら、サーチしたい範囲を選択する。範囲の選択方法は、まず出発点でマウスの左ボタンを押し、そのまま終点までドラッグすると、それまでの範囲が四角形で示されるので適当なところでボタンを離す。元の範囲に戻したいときはマウスの右ボタンをクリックする。(この機能は、PatViewのどのモードでも有効に働く)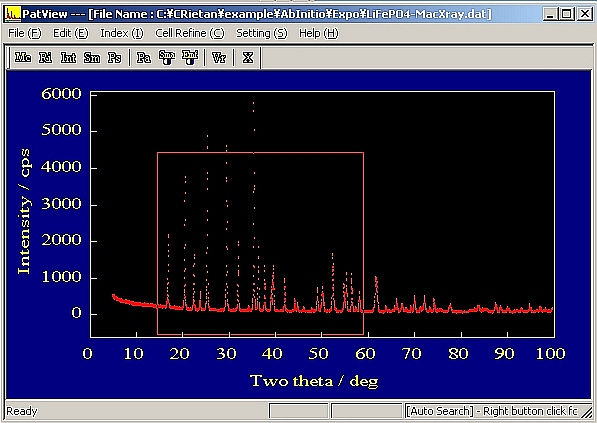
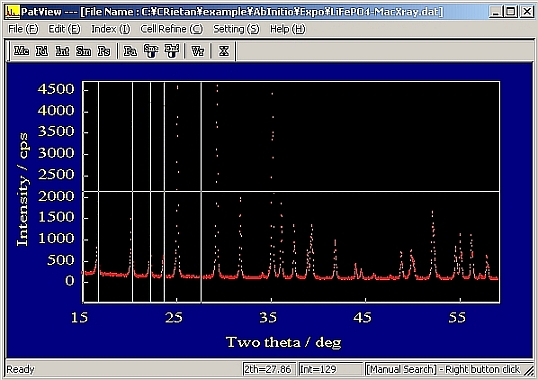
- 範囲を選択した状態で、[Edit]->[Manual Peak Search]を実行。マウスのボタンを離したままで、マウスを移動させると十字のラインが動く。この状態でピークトップの位置で左ダブルクリックするとその位置にマークされる。画面上の見た目のピークトップと実際のピークトップとは若干ずれているようなので、下の枠のステイタスバーのところに2θと強度が表示されるのでこれに見ながら左ダブルクリックするとよい。
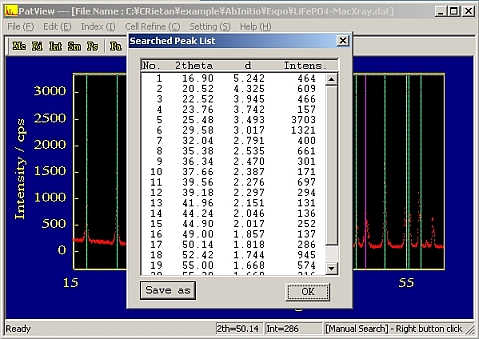
- 次にこのモードのままで[Edit]->[Searched Peak List]を実行すると、それまでマークされたピークのリストが表示される。このデータを保存したければ[Save As]で適当なファイルに保存できる。
- サーチしたピークリストを用いて指数付けを行うには、後述する「ITO」や「Treor」などのプログラムを用いることになるが、これらのプログラムの使い方は結構ややこしい。Auto Searchでは、これらのプログラムをPatViewから直接呼び出して指数付けを、更に格子定数の精密化行うことができるようになっているので、これ以降のやり方はそちらを参照。
回折データを読み込んだら、Manual Searchと同様にサーチしたい範囲を選択する。
- [Edit]->[Auto Peak Search]を実行すると、[Shreshould]の値を聞いてくるので、適当な値を代入する。この値の意味は、この値以上のピーク強度を持つものをサーチするということ。つづいて[Eliminate Ka2 Peaks]と聞いてくるので通常はOKを押してKα2に起因するピークを除外する。更に[Conditions for Ka2 Elimination]を聞いてくる。2つのTolerance Factorはデフォルトのままで大抵よい。[OK}を押すと見つけたピーク位置にバーが示される。
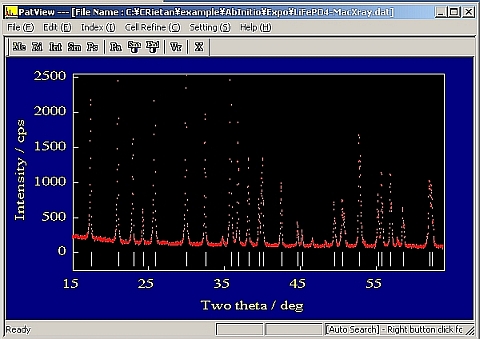
- ピークの数が少なすぎたり、多すぎたりしたら、[Shreshould]の値を変更して2の操作を繰り返す。
- ピークがほぼ見つかったら、このモードのままで[Edit]->[Searched Peak List]を実行すると、それまでマークされたピークのリストが表示される。このデータを保存したければ[Save As]で適当なファイルに保存できる。
- 引き続き指数付けのプロセスに移行したければ、このままのモードで次の指数付けを行う。これで終了したければ、右クリックでこのモードから抜け出す。
指数付けのプログラムはアカデミックユーザに対してフリーな「ITO」、「Treor」、「Dicvol」および「Cell」の4つのプログラムがPatViewから直接実行できる。それぞれのプログラムの使い方はPatViewをインストールしたディレクトリにあるマニュアルを参照してください。しかし、このPatViewではこれらプログラムの詳細な使用方法を知らなくても簡単に指数付けを行えるようにしてある。
- 前述の5の操作から引き続き指数付けに入るには、このモードのままで[Index]->[Ito Run]を実行する。ItoのところはTreor又はDicvolでもよい。
- 「Do you edit input file ?」と聞いてくるが、これらのプログラムの使用法に詳しければ「OK]を押して自分で適当に修正してもかまわない。大抵は[No]で解が求められる。
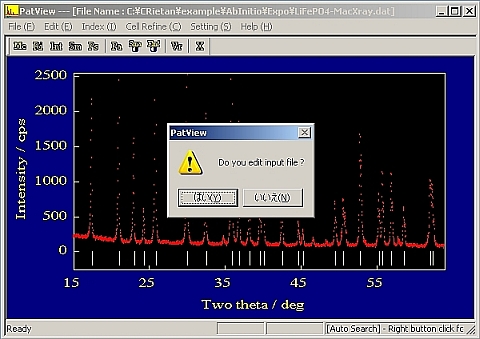
- 大抵の場合、一瞬で計算が完了してアウトプットファイルが表示される。大変詳細な情報が書き込まれているのでじっくり検討することが望ましい。しかし早く結果が見たい人はエディタを終了して、[Cell Refine]->[Prepare from Ito](ItoのところはTreor又はDicvolでもよい)を実行すると、エディタが立ち上がる。このファイルは後述の「CellRef」の入力ファイルである。この例ではOrthorhombicで指数付けされた。
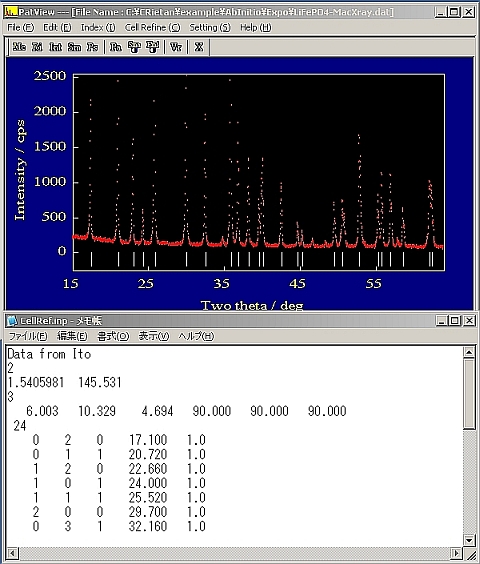
- この段階で指数付けに失敗していると、入力ファイルの作成に失敗した旨のメッセージが表示される。この場合には、Auto Searchからやり直す必要がある。また、複数の解が見つかった場合には、そのメッセージが表示されて「OK」を押すと、複数の解が新しいウィンドウ上に表示されるので、適当な候補の番号を入力してこのウィンドウを閉じる。
- 格子定数の精密化プログラム「CellRef」用のインプットファイルがITO出力ファイルから作成される。書いてある内容は殆ど理解できるはずであるが、ファイルの最後の方に指数付けできなかったピークがリストされることがある。問題がなければエディタを終了する。
- 再びPatViewに戻ってから[Cell Refine]->[CellRef Run]を実行する。CellRef用に必要な入力データは既に入力されているので、[Refine]->[Execute]を実行すると精密化された格子定数が表示される。
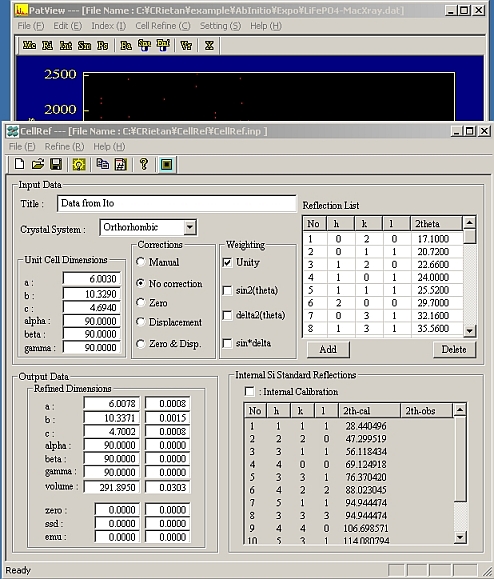
- CellRefの詳しい使用方法は「CellRef.txt」を参照。
- Cellの使い方は次の[空間群の探索]を参照。
注意:
指数付けプログラムの中で「Dicvol」は殆ど役に立たない。また、指数付けプログラムとして、純国産の「Cell」(大阪教育大の故高木先生開発)がある。このプログラムは優れもので、空間群の候補まで予測してくれる。ただし、このプログラムは結晶系を決めておかないと使用できない。
- [Index]->[Cell Run]でCellが起動する。
- メニュー1の[Program List and Execution]を選択する。
- CellのメニューからITOやTreor等で示された結晶系のプログラムを選択する。この場合は、CELLO(Orthorhombic)を選択する。
- 入力ファイルは”Cell”を選択する。
- 種々の設定パラメータを聞いてくるが、Default値でよい。全てリターンでDefault値を入力すると計算が始まる。
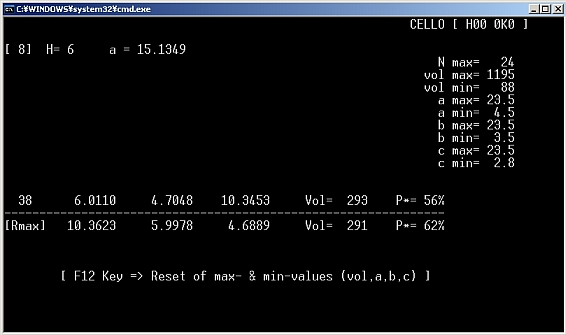
- 指数付けができ、結晶系が推定されたら上記のCellを用いて再び指数付けを行うと、格子定数と空間群を予想してくれる。Cellは複数の格子定数の候補を示してくれるが、ITOやTreorが示した格子定数とほぼ一致していれば信頼できる。
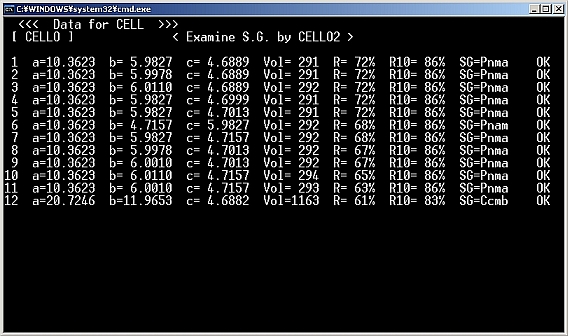
- Cellが示す空間群は、観測された指数の消滅則を満足するものであるが、注意しなければならないことは、同じ消滅則の空間群は複数ある場合があることである。どれが正しい空間群であるかについては、その後のRietveld解析や、類似の化合物の空間群を参考にしなければ決定できない。この例ではITOが予想したOrthorhombicで、空間群は非常に高い信頼性でPnmaを予想している。
- Cellを抜けるときにCellの出力ファイルが示されるので、適当な名前で保存しておくとよい。
- [Index]->[Prepare From EXPO File From Cell]でEXPOの入力ファイルを作る。
- Cellが予想した空間群がたくさん示されるが、選択する一応の目安は%の値が大きいもの。
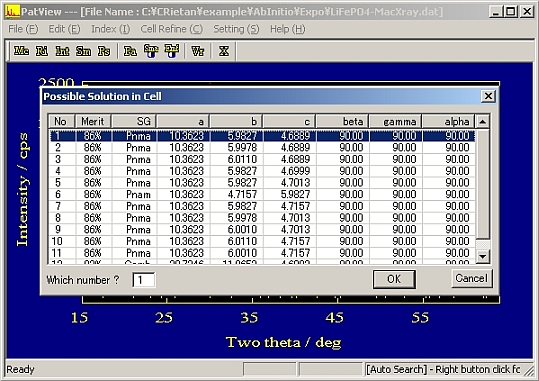
- 新しいWindowが開き、空欄の項目を書き込む。
入力する項目
・タイトル
・EXPO用の粉末データ名 −−−絶対パス+ファイル名で拡張子は*.int、このファイルはRietan2000でそのまま使用できる。
・化学式 −−−単位格子中に含まれる全原子の個数、NaClでZ=8ならば”Na 8 Cl 8"と記入する。
・空間群 −−−右側にCellで予想された空間群が示されるので、まずはこれを記入する。(空白の入れ方に注意、この場合は、P n m a)
・その他は、default値でよい。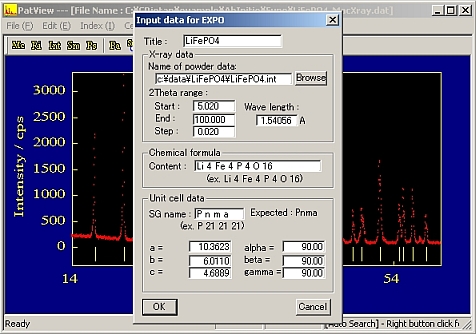
- [Ok]を押してWindowを閉じる。
5.6 EXPOの実行
- EXPOの実行はEXPOのマニュアル参照すること。
- メニューから前述のEXPOの入力ファイルを選択する。[User working directory]に正確に記入し、[Browse]を押すと、Expoの入力ファイルのリストが現れるので先ほど作成したファイルを選択する。
- EXPOの実行画面上に粉末パターンが現れたら、基本的には[continue]と[Ok]を押し続けること。
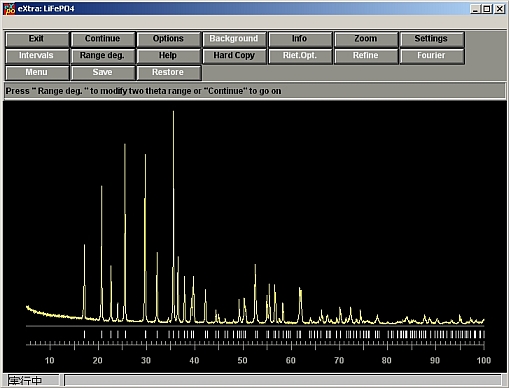
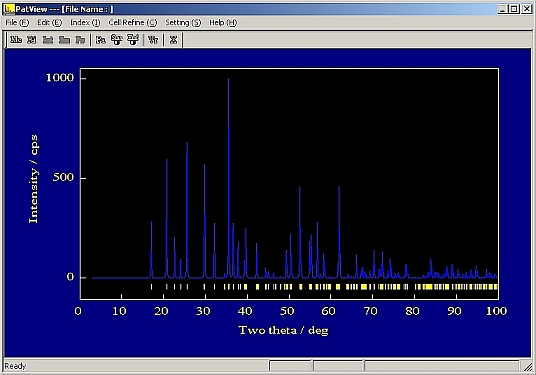
- 1つの解だけではなく10個くらいの解を出すこと。下の図は数回繰り返し解を求めたところ。
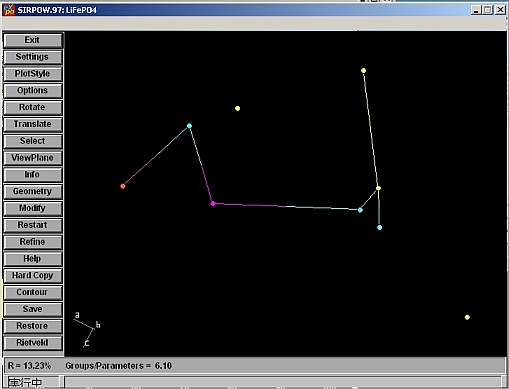
- R因子が10%台の解が1つでも見つかれば、構造が解けた可能性が高い。
- EXPOを終了して、PatViewの画面に戻る。
5.7 EXPOの解からRIetan2000用の入力ファイルの作成
- [Index]->[GenHKL Run]を実行する。
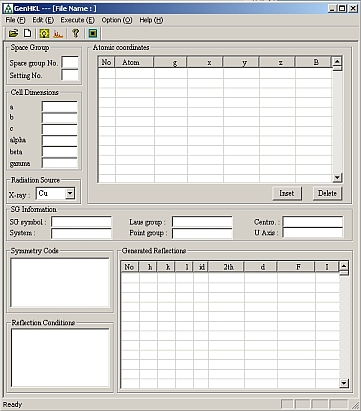
- [File]->[File Open]から、*.outを選択してから先のEXPOの出力ファイルを開く。
- EXPOが見つけた構造のリストが表示されるので、適当な解を選択する。
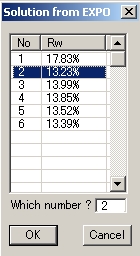
- まず、EXPOの出力ファイルには空間群番号がないので、[Edit]->[SG Number]を実行すると、空間群と設定番号を見つけてくれる。座標リストの中に”Q”の表示があったらこれを削除する。また、余分な原子があったらこれも削除する。(どれが余分な原子かを判断するのは結構難しい)。下図はリストの最後の3つのLiを削除してStructViewで構造を見たところ。実際これが正解の構造。
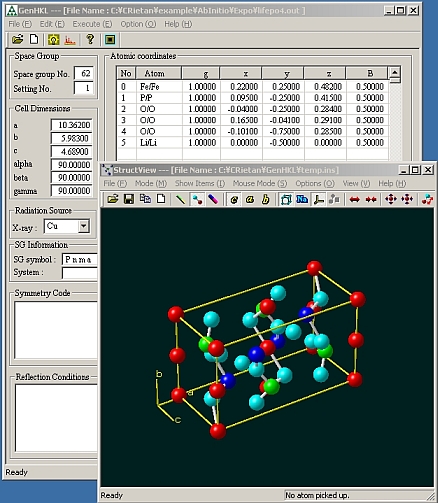
- [Execute]->[Calculate Profile]を実行し、シミュレートしたパターンと実測パターを比較する。この段階で、実測とあまりあわない場合は、EXPOの他の解を選択して同様の操作を繰り返す。
- パターンが大体合っていれば、[Execute]->[Show Structure]を実行し、構造を見る。大抵の場合、結晶学的におかしい原子の並びになっている。この段階で、PやSなどがあればPO4やSO4などの四面体の構造などが見えてくることがある。余分な原子もリストアップされている場合が多いので、それらを削除して、妥当な構造になるように原子の追加と削除を繰り返すとよい。下図はシミュレートしたパターンで、実測のパターンと大体一致している。
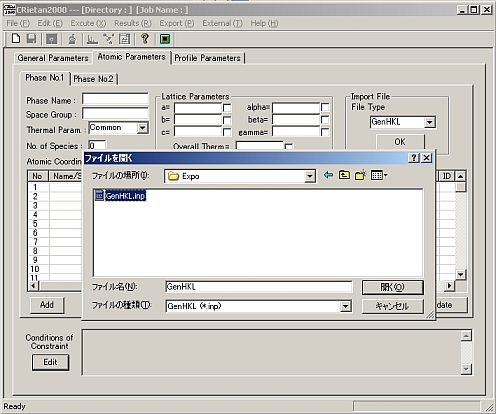
- ほぼ妥当な構造になったら、この結果をGenHKLのインプットファイルとして適当なdirectoryに保存する。このファイルはCRietan2000から読み込むことができるので、リートベルド解析を行うことができる。
- 下図はCRietan2000の[Atomic Parameter]の[Import File]でGenHKLファイルを選択して先に作成したGenHKL.inoファイルを読み込んでいるところ。